Publications
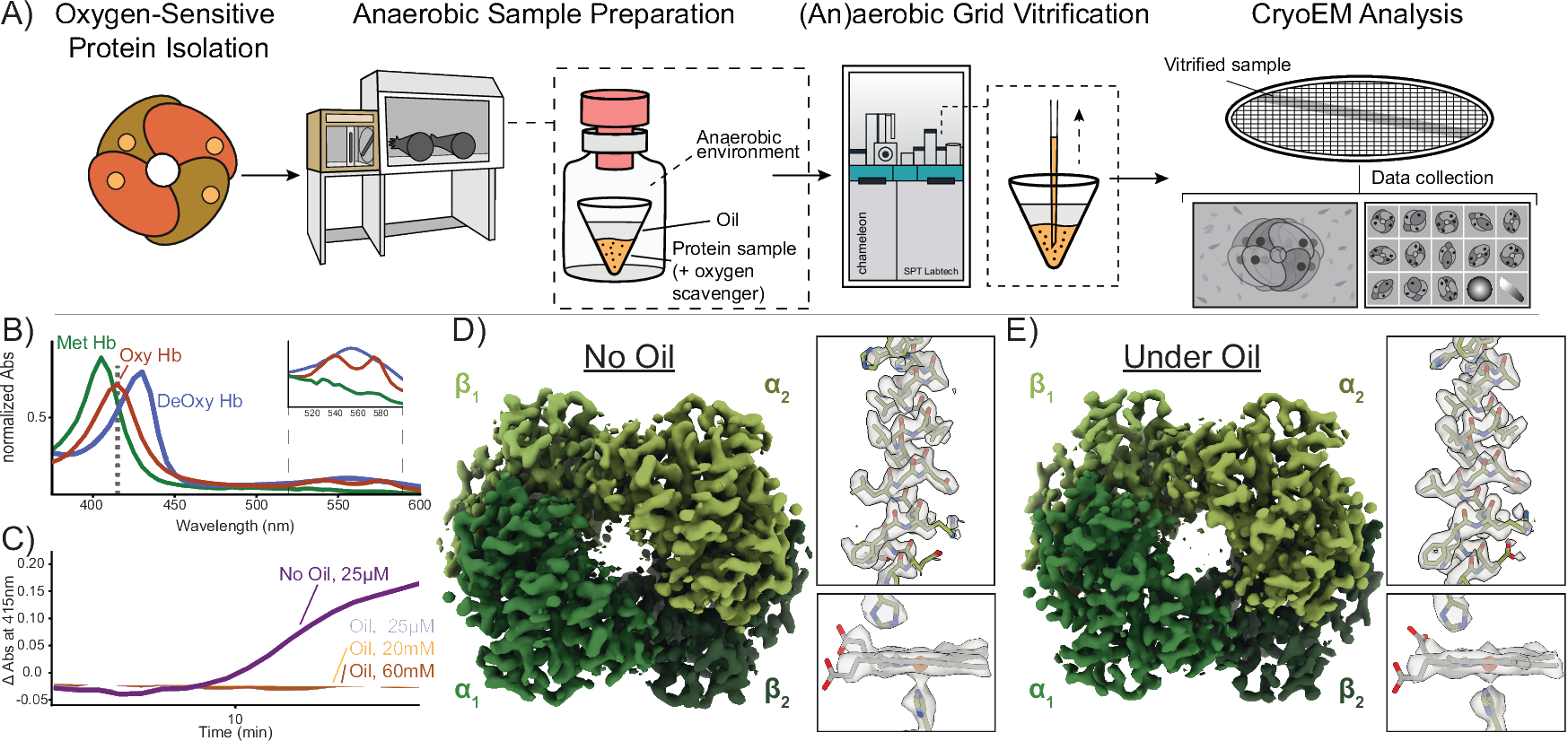
Preparation of oxygen-sensitive proteins for high-resolution cryoEM structure determination using blot-free vitrification
Nature Communications
2025
Cook BD*, Narehood SM*, McGuire KL*, Li Y, Tezcan FA, Herzik MA Jr.
Abstract
*These authors contributed equally
High-quality grid preparation for single-particle cryogenic electron microscopy (cryoEM) remains a bottleneck for routinely obtaining high-resolution structures. The issues that arise from traditional grid preparation workflows are particularly exacerbated for oxygen-sensitive proteins, including metalloproteins, whereby oxygen-induced damage and alteration of oxidation states can result in protein inactivation, denaturation, and/or aggregation. Indeed, 99% of the current structures in the EMBD were prepared aerobically and limited successes for anaerobic cryoEM grid preparation exist. Current practices for anaerobic grid preparation involve a vitrification device located in an anoxic chamber, which presents significant challenges including temperature and humidity control, optimization of freezing conditions, costs for purchase and operation, as well as accessibility. Here, we present a streamlined approach that allows for the (an)aerobic vitrification of oxygen-sensitive proteins using an automated aerobic blot-free grid vitrification device - the SPT Labtech chameleon. This robust workflow allows for high-resolution structure determination of dynamic, oxygen-sensitive proteins, of varying complexity and molecular weight.
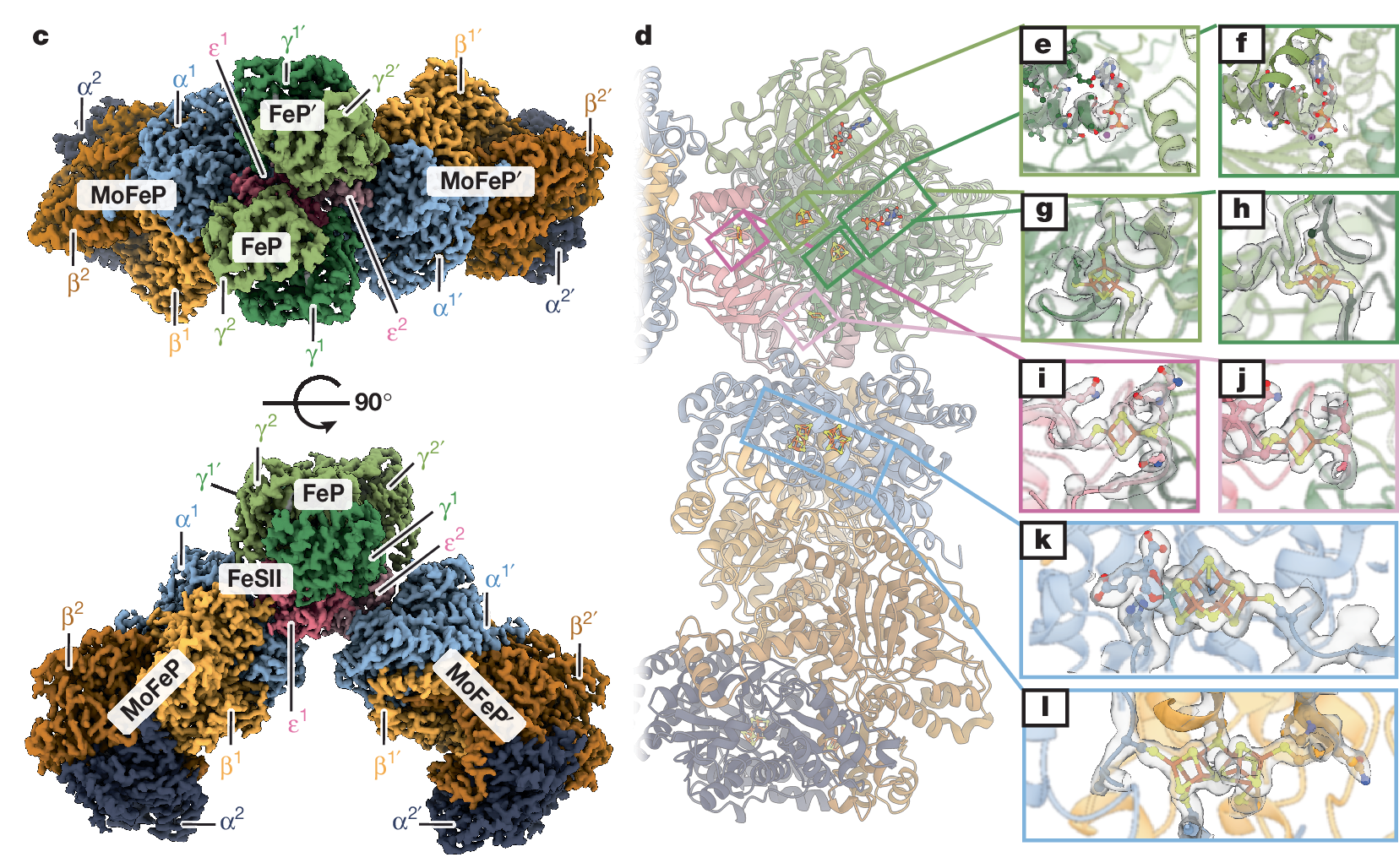
Structural basis for the conformational protection of nitrogenase from O₂
Nature
2025
Narehood SM, Cook BD, Srisantitham S, Eng VH, Shiau AA, McGuire KL, Britt RD, Herzik MA Jr, Tezcan FA
Abstract
The low reduction potentials required for the reduction of dinitrogen (N2) render metal-based nitrogen-fxation catalysts vulnerable to irreversible damage by dioxygen (O2). Such O2 sensitivity represents a major conundrum for the enzyme nitrogenase, as a large fraction of nitrogen-fxing organisms are either obligate aerobes or closely associated with O2-respiring organisms to support the high energy demand of catalytic N2 reduction. To counter O2 damage to nitrogenase, diazotrophs use O2 scavengers, exploit compartmentalization or maintain high respiration rates to minimize intracellular O2 concentrations. A last line of damage control is provided by the 'conformational protection' mechanism, in which a [2Fe:2S] ferredoxin-family protein termed FeSII (ref. 10) is activated under O2 stress to form an O2-resistant complex with the nitrogenase component proteins. Despite previous insights, the molecular basis for the conformational O2 protection of nitrogenase and the mechanism of FeSII activation are not understood. Here we report the structural characterization of the Azotobacter vinelandii FeSII-nitrogenase complex by cryo-electron microscopy. Our studies reveal a core complex consisting of two molybdenum-iron proteins (MoFePs), two iron proteins (FePs) and one FeSII homodimer, which polymerize into extended flaments. In this three-protein complex, FeSII mediates an extensive network of interactions with MoFeP and FeP to position their iron-sulphur clusters in catalytically inactive but O2-protected states. The architecture of the FeSII-nitrogenase complex is confrmed by solution studies, which further indicate that the activation of FeSII involves an oxidation-induced conformational change.
Access the Paper
- PMID: 39779844
- PMCID: PMC11812610
Additional Links
- PDBs: 9CTZ 9CU0 9CU1 9CU2
- EMDBs: EMD-45923 EMD-45924 EMD-45925 EMD-45926
- Tezcan Lab
- Herzik Lab
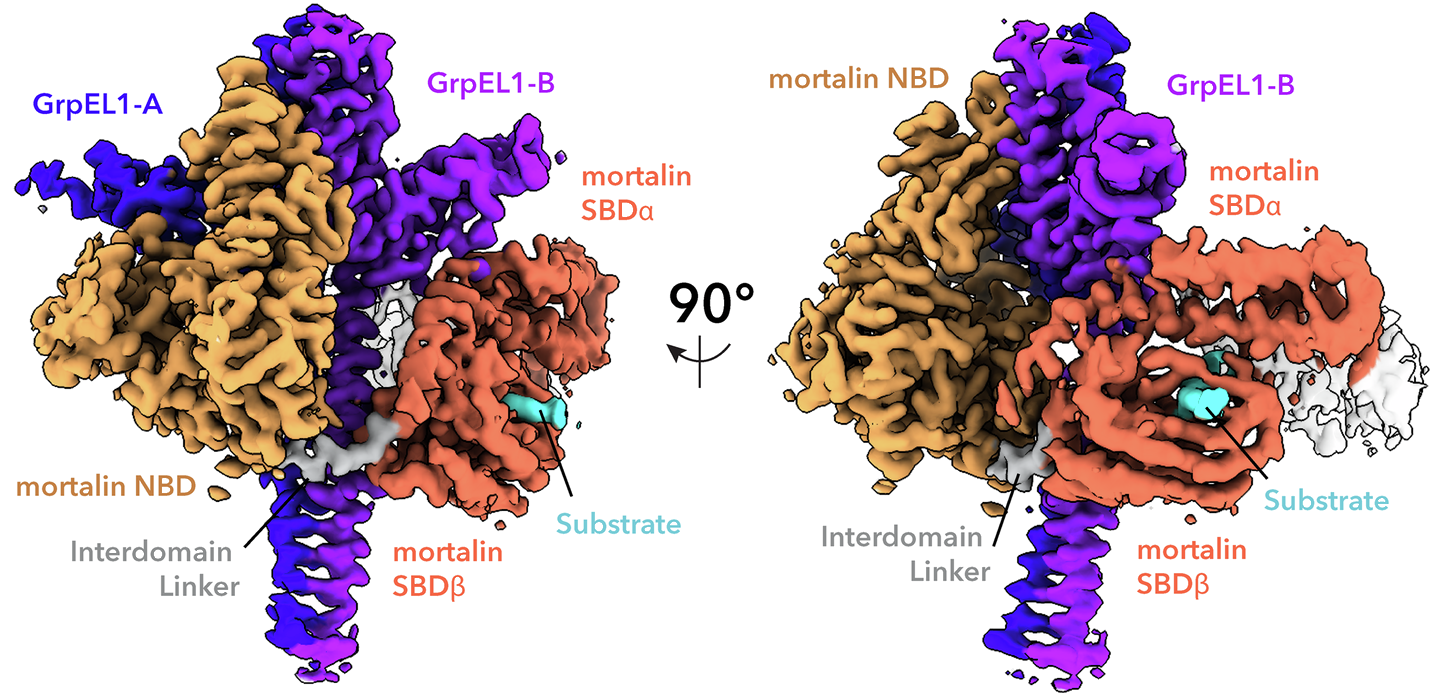
Structural insights into GrpEL1-mediated nucleotide and substrate release of human mitochondrial Hsp70
Nature Communications
2024
Morizono M, McGuire KL, Birouty N, Herzik MA Jr.
Abstract
Maintenance of protein homeostasis is necessary for cell viability and depends on a complex network of chaperones and co-chaperones, including the heat-shock protein 70 (Hsp70) system. In human mitochondria, mitochondrial Hsp70 (mortalin) and the nucleotide exchange factor (GrpEL1) work synergistically to stabilize proteins, assemble protein complexes, and facilitate protein import. However, our understanding of the molecular mechanisms guiding these processes is hampered by limited structural information. To elucidate these mechanistic details, we used cryoEM to determine the first structures of full-length human mortalin-GrpEL1 complexes in previously unobserved states. Our structures and molecular dynamics simulations allow us to delineate specific roles for mortalin-GrpEL1 interfaces and to identify steps in GrpEL1-mediated nucleotide and substrate release by mortalin. Subsequent analyses reveal conserved mechanisms across bacteria and mammals and facilitate a complete understanding of sequential nucleotide and substrate release for the Hsp70 chaperone system.

Machine learning approaches to cryoEM density modification differentially affect biomacromolecule and ligand density quality
Frontiers in Molecular Biosciences
2024
Berkeley RF*, Cook BD*, Herzik MA Jr.*
Abstract
*These authors contributed equally
The application of machine learning to cryogenic electron microscopy (cryoEM) data analysis has added a valuable set of tools to the cryoEM data processing pipeline. As these tools become more accessible and widely available, the implications of their use should be assessed. We noticed that machine learning map modification tools can have differential effects on cryoEM densities. In this perspective, we evaluate these effects to show that machine learning tools generally improve densities for biomacromolecules while generating unpredictable results for ligands. This unpredictable behavior manifests both in quantitative metrics of map quality and in qualitative investigations of modified maps. The results presented here highlight the power and potential of machine learning tools in cryoEM, while also illustrating some of the risks of their unexamined use.
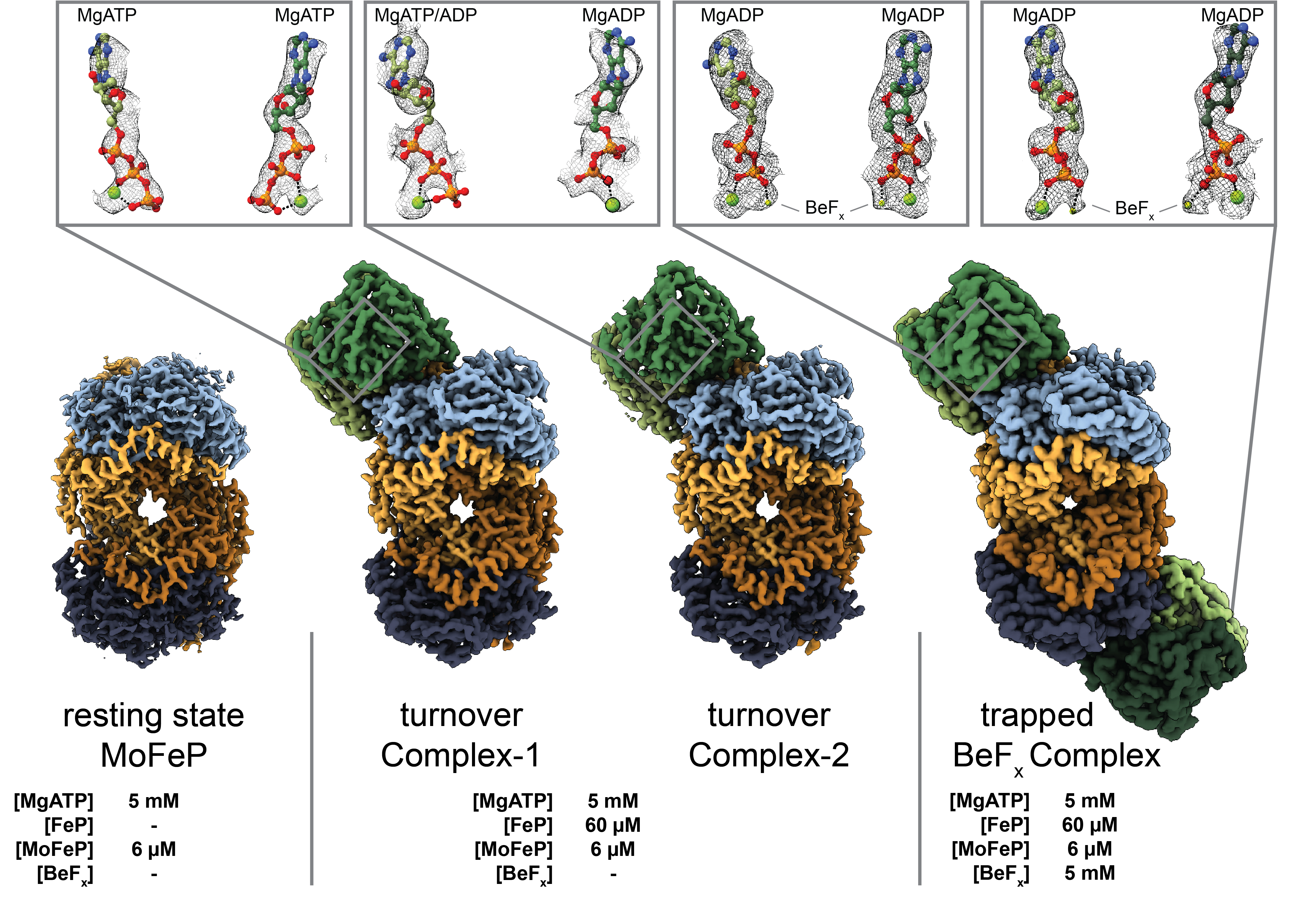
Structures of the nitrogenase complex prepared under catalytic turnover conditions
Science
2022
Rutledge HL, Cook BD, Nguyen HPM, Herzik MA Jr., Tezcan FA
Abstract
The enzyme nitrogenase couples adenosine triphosphate (ATP) hydrolysis to the multielectron reduction of atmospheric dinitrogen into ammonia. Despite extensive research, the mechanistic details of ATP-dependent energy transduction and dinitrogen reduction by nitrogenase are not well understood, requiring new strategies to monitor its structural dynamics during catalytic action. Here, we report cryo-electron microscopy structures of the nitrogenase complex prepared under enzymatic turnover conditions. We observe that asymmetry governs all aspects of the nitrogenase mechanism, including ATP hydrolysis, protein-protein interactions, and catalysis. Conformational changes near the catalytic iron-molybdenum cofactor are correlated with the nucleotide-hydrolysis state of the enzyme.
Manual Blot-and-Plunge Freezing of Biological Specimens for Single-Particle Cryogenic Electron Microscopy
Journal of Visualized Experiments
2022
Nguyen HMP*, McGuire KL*, Cook BD*, Herzik MA Jr.
Abstract
*These authors contributed equally
Imaging biological specimens with electrons for high-resolution structure determination by single-particle cryogenic electron microscopy (cryoEM) requires a thin layer of vitreous ice containing the biomolecules of interest. Despite numerous technological advances in recent years that have propelled single-particle cryoEM to the forefront of structural biology, the methods by which specimens are vitrified for high-resolution imaging often remain the rate-limiting step. Although numerous recent efforts have provided means to overcome hurdles frequently encountered during specimen vitrification, including the development of novel sample supports and innovative vitrification instrumentation, the traditional manually operated plunger remains a staple in the cryoEM community due to the low cost to purchase and ease of operation. Here, we provide detailed methods for using a standard, guillotine-style manually operated blot-and-plunge device for the vitrification of biological specimens for high-resolution imaging by single-particle cryoEM. Additionally, commonly encountered issues and troubleshooting recommendations for when a standard preparation fails to yield a suitable specimen are also described.
Cryo-EM model validation recommendations based on outcomes of the 2019 EMDataResource challenge
Nature Methods
2021
Lawson CL, Kryshtafovych A, Adams PD, Afonine PV, Baker ML, Barad BA, Bond P, Burnley T, Cao R, Cheng J, Chojnowski G, Cowtan K, Dill KA, DiMaio F, Farrell DP, Fraser JS, Herzik MA, Hoh SW, Hou J, Hung L, Igaev M, Joseph AP, Kihara D, Kumar D, Mittal S, Monastyrskyy B, Olek M, Palmer CM, Patwardhan A, Perez A, Pfab J, Pintilie GD, Richardson JS, Rosenthal PB, Sarkar D, Schäfer LU, Schmid MF, Schröder GF, Shekhar M, Si D, Singharoy A, Terashi G, Terwilliger TC, Vaiana A, Wang L, Wang Z, Wankowicz SA, Williams CJ, Winn M, Wu T, Yu X, Zhang K, Berman HM, Chiu W
Abstract
This paper describes outcomes of the 2019 Cryo-EM Model Challenge. The goals were to (1) assess the quality of models that can be produced from cryogenic electron microscopy (cryo-EM) maps using current modeling software, (2) evaluate reproducibility of modeling results from different software developers and users and (3) compare performance of current metrics used for model evaluation, particularly Fit-to-Map metrics, with focus on near-atomic resolution. Our findings demonstrate the relatively high accuracy and reproducibility of cryo-EM models derived by 13 participating teams from four benchmark maps, including three forming a resolution series (1.8 to 3.1 Å). The results permit specific recommendations to be made about validating near-atomic cryo-EM structures both in the context of individual experiments and structure data archives such as the Protein Data Bank. We recommend the adoption of multiple scoring parameters to provide full and objective annotation and assessment of the model, reflective of the observed cryo-EM map density.
Sub-2 Angstrom resolution structure determination using single-particle cryo-EM at 200 keV
Journal of Structural Biology: X
2020
Wu M, Lander GC, Herzik MA Jr.
Abstract
Although the advent of direct electron detectors (DEDs) and software developments have enabled the routine use of single-particle cryogenic electron microscopy (cryo-EM) for structure determination of well-behaved specimens to high-resolution, there nonetheless remains a discrepancy between the resolutions attained for biological specimens and the information limits of modern transmission electron microscopes (TEMs). Instruments operating at 300 kV equipped with DEDs are the current paradigm for high-resolution single-particle cryo-EM, while 200 kV TEMs remain comparatively underutilized for purposes beyond sample screening. Here, we expand upon our prior work and demonstrate that one such 200 kV microscope, the Talos Arctica, equipped with a K2 DED is capable of determining structures of macromolecules to as high as ~1.7 Å resolution. At this resolution, ordered water molecules are readily assigned and holes in aromatic residues can be clearly distinguished in the reconstructions. This work emphasizes the utility of 200 kV electrons for high-resolution single-particle cryo-EM and applications such as structure-based drug design.
Access the Paper
- PMID: 32647824
- PMCID: PMC7337053
Additional Links
- PDBs: 6V20 6V21
- EMDBs: EMD-21023 EMD-21024
- Raw Data: EMPIAR-10337, EMPIAR-10338, EMPIAR-
- Herzik Lab
- Lander Lab
High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM
Nature Communications
2019
Herzik MA. Jr.*, Wu M*, Lander GC
Abstract
*These authors contributed equally
Determining high-resolution structures of biological macromolecules with masses of less than 100 kilodaltons (kDa) has long been a goal of the cryo-electron microscopy (cryo-EM) community. While the Volta Phase Plate has enabled cryo-EM structure determination of biological specimens of this size range, use of this instrumentation is not yet fully automated and can present technical challenges. Here, we show that conventional defocus-based cryo-EM methodologies can be used to determine the high-resolution structures of specimens amassing less than 100 kDa using a transmission electron microscope operating at 200 keV coupled with a direct electron detector. Our ~2.9 Å structure of alcohol dehydrogenase (82 kDa) proves that bound ligands can be resolved with high fidelity, indicating that these methodologies can be used to investigate the molecular details of drug-target interactions. Our ~2.8 Å and ~3.2 Å resolution structures of methemoglobin demonstrate that distinct conformational states can be identified within a dataset for proteins as small as 64 kDa. Furthermore, we provide the first sub-nanometer cryo-EM structure of a protein smaller than 50 kDa.
Access the Paper
- PMID: 30833564
- PMCID: PMC6399227
- Biorxiv Preprint: 489898
Additional Links
- PDBs: 6NBB 6NBC 6NBD
- EMDBs: EMD-0406 EMD-0407 EMD-0408
- Raw Data: EMPIAR-10249, EMPIAR-10250, EMPIAR-10252
- Lander Lab
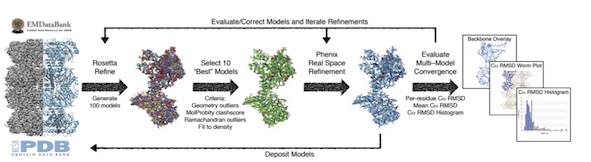
A Multi-model Approach to Assessing Local and Global Cryo-EM Map Quality
Structure
2019
Herzik MA Jr., Fraser. JS, Lander, GC
Abstract
There does not currently exist a standardized indicator of how well a cryo-EM-derived model represents the density from which it was generated. We present a straightforward methodology that utilizes freely available tools to generate a suite of independent models and to evaluate their convergence in an EM density. These analyses provide both a quantitative and qualitative assessment of the precision of the models and their representation of the density, respectively, while concurrently providing a platform for assessing both global and local EM map quality. We further use standardized datasets to provide an expected model-model agreement criterion for EM maps reported to be at 5 Å resolution or better. Associating multiple atomic models with a deposited EM map provides a rapid and accessible reporter of convergence, a strong indicator of highly resolved molecular detail, and is an important step toward an FSC-independent assessment of map and model quality.
Access the Paper
- PMID: 30449687
- PMCID: PMC6365196
- Biorxiv Preprint: 128561
Additional Links
- PDBs:
- EMDBs:
- Raw Data: EMPIAR-, EMPIAR-, EMPIAR-
- Fraser Lab
- Lander Lab
Conformational ensemble of the human TRPV3 ion channel
Nature Communications
2018
Zubcevic L*, Herzik MA Jr*, Wu M*, Borschel WF, Hirschi M, Song AS, Lander GC, Lee SY.
Abstract
*These authors contributed equally
Transient receptor potential vanilloid channel 3 (TRPV3), a member of the thermosensitive TRP (thermoTRPV) channels, is activated by warm temperatures and serves as a key regulator of normal skin physiology through the release of pro-inflammatory messengers. Mutations in trpv3 have been identified as the cause of the congenital skin disorder, Olmsted syndrome. Unlike other members of the thermoTRPV channel family, TRPV3 sensitizes upon repeated simulation, yet a lack of structural information about the channel precludes a molecular-level understanding of TRPV3 sensitization and gating. Here, we present the cryo-electron microscopy structures of apo and sensitized human TRPV3, as well as several structures of TRPV3 in the presence of the common thermoTRPV agonist 2-aminoethoxydiphenyl borate (2-APB). Our results show α-to-Φ-helix transitions in the S6 during sensitization, and suggest a critical role for the S4-S5 linker Φ-helix during ligand gating.
Cryo-EM structure of a mitochondrial calcium uniporter
Science
2018
Yoo J*, Wu M*, Yin Y, Herzik MA Jr, Lander GC, Lee SY.
Abstract
*These authors contributed equally
Calcium transport plays an important role in regulating mitochondrial physiology and pathophysiology. The mitochondrial calcium uniporter (MCU) is a calcium-selective ion channel that is the primary mediator for calcium uptake into the mitochondrial matrix. Here, we present the cryo-electron microscopy structure of the full-length MCU from Neurospora crassa to an overall resolution of ~3.7 angstroms. Our structure reveals a tetrameric architecture, with the soluble and transmembrane domains adopting different symmetric arrangements within the channel. The conserved W-D-Φ-Φ-E-P-V-T-Y sequence motif of MCU pore forms a selectivity filter comprising two acidic rings separated by one helical turn along the central axis of the channel pore. The structure combined with mutagenesis gives insight into the basis of calcium recognition.
Structural and cellular mechanisms of peptidyl-prolyl isomerase Pin1-mediated enhancement of Tissue Factor gene expression, protein half-life, and pro-coagulant activity
Haematologica
2018
Kurakula K, Koenis DS, Herzik MA Jr, Liu Y, Craft JW Jr, van Loenen PB, Vos M, Tran MK, Versteeg HH, Goumans MTH, Ruf W, de Vries CJM, Şen M.
Abstract
Tissue Factor is a cell-surface glycoprotein expressed in various cells of the vasculature and is the principal regulator of the blood coagulation cascade and hemostasis. Notably, aberrant expression of Tissue Factor is associated with cardiovascular pathologies such as atherosclerosis and thrombosis. Here, we sought to identify factors that regulate Tissue Factor gene expression and activity. Tissue Factor gene expression is regulated by various transcription factors, including activating protein-1 and nuclear factor-κ B. The peptidyl-prolyl isomerase Pin1 is known to modulate the activity of these two transcription factors, and we now show that Pin1 augments Tissue Factor gene expression in both vascular smooth muscle cells and activated endothelial cells via activating protein-1 and nuclear factor-κ B signaling. Furthermore, the cytoplasmic domain of Tissue Factor contains a well-conserved phospho-Ser258-Pro259 amino-acid motif recognized by Pin1. Using co-immunoprecipitation and solution nuclear magnetic resonance spectroscopy, we show that the WW-domain of Pin1 directly binds the cytoplasmic domain of Tissue Factor. This interaction occurs via the phospho-Ser258-Pro259 sequence in the Tissue Factor cytoplasmic domain and results in increased protein half-life and pro-coagulant activity. Taken together, our results establish Pin1 as an upstream regulator of Tissue Factor-mediated coagulation, thereby opening up new avenues for research into the use of specific Pin1 inhibitors for the treatment of diseases characterized by pathological coagulation, such as thrombosis and atherosclerosis.
Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3
Nature
2017
Hirschi M*, Herzik MA Jr*, Wie J, Suo Y, Borschel WF, Ren D, Lander GC, Lee SY.
Abstract
*These authors contributed equally
The modulation of ion channel activity by lipids is increasingly recognized as a fundamental component of cellular signalling. The transient receptor potential mucolipin (TRPML) channel family belongs to the TRP superfamily and is composed of three members: TRPML1-TRPML3. TRPMLs are the major Ca2+-permeable channels on late endosomes and lysosomes (LEL). They regulate the release of Ca2+ from organelles, which is important for various physiological processes, including organelle trafficking and fusion. Loss-of-function mutations in the MCOLN1 gene, which encodes TRPML1, cause the neurodegenerative lysosomal storage disorder mucolipidosis type IV, and a gain-of-function mutation (Ala419Pro) in TRPML3 gives rise to the varitint-waddler (Va) mouse phenotype. Notably, TRPML channels are activated by the low-abundance and LEL-enriched signalling lipid phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2), whereas other phosphoinositides such as PtdIns(4,5)P2, which is enriched in plasma membranes, inhibit TRPMLs. Conserved basic residues at the N terminus of the channel are important for activation by PtdIns(3,5)P2 and inhibition by PtdIns(4,5)P2. However, owing to a lack of structural information, the mechanism by which TRPML channels recognize PtdIns(3,5)P2 and increase their Ca2+ conductance remains unclear. Here we present the cryo-electron microscopy (cryo-EM) structure of a full-length TRPML3 channel from the common marmoset (Callithrix jacchus) at an overall resolution of 2.9 Å. Our structure reveals not only the molecular basis of ion conduction but also the unique architecture of TRPMLs, wherein the voltage sensor-like domain is linked to the pore via a cytosolic domain that we term the mucolipin domain. Combined with functional studies, these data suggest that the mucolipin domain is responsible for PtdIns(3,5)P2 binding and subsequent channel activation, and that it acts as a 'gating pulley' for lipid-dependent TRPML gating.
Achieving better-than-3-Å resolution by single-particle cryo-EM at 200 keV
Nature Methods
2017
Herzik MA Jr*, Wu M*, Lander GC
Abstract
*These authors contributed equally
Technical and methodological advances in single-particle cryo-electron microscopy (cryo-EM) have expanded the technique into a resolution regime that was previously only attainable by X-ray crystallography. Although single-particle cryo-EM has proven to be a useful technique for determining the structures of biomedically relevant molecules at near-atomic resolution, nearly 98% of the structures resolved to better than 4 Å resolution have been determined using 300 keV transmission electron microscopes (TEMs). We demonstrate that it is possible to obtain cryo-EM reconstructions of macromolecular complexes at a range of sizes to better than 3 Å resolution using a 200 keV TEM. These structures are of sufficient quality to unambiguously assign amino acid rotameric conformations and identify ordered water molecules, features previously thought only to be resolvable using TEMs operating at 300 keV.
Access the Paper
- PMID: 28991891
- PMCID: PMC5679434
- Biorxiv Preprint: 141994
Additional Links
- PDBs: 5VY3 5VY4 5VY5
- EMDBs: EMD-8741 EMD-8742 EMD-8743
- Raw Data: EMPIAR-10181, EMPIAR-10185, EMPIAR-10186
- Lander Lab
Regulation of nitric oxide signaling by formation of a distal receptor-ligand complex
Nature Chemical Biology
2017
Guo Y, Suess DLM, Herzik MA Jr, Iavarone AT, Britt RD, Marletta MA.
Abstract
The binding of nitric oxide (NO) to the heme cofactor of heme-nitric oxide/oxygen binding (H-NOX) proteins can lead to the dissociation of the heme-ligating histidine residue and yield a five-coordinate nitrosyl complex, an important step for NO-dependent signaling. In the five-coordinate nitrosyl complex, NO can reside on either the distal or proximal side of the heme, which could have a profound influence over the lifetime of the in vivo signal. To investigate this central molecular question, we characterized the Shewanella oneidensis H-NOX (So H-NOX)-NO complex biophysically under limiting and excess NO conditions. The results show that So H-NOX preferably forms a distal NO species with both limiting and excess NO. Therefore, signal strength and complex lifetime in vivo will be dictated by the dissociation rate of NO from the distal complex and the rebinding of the histidine ligand to the heme.
Access the Paper
- PMID: 28967923
- PMCID: PMC5698159
Additional Links
- PDBs:
- EMDBs:
- Raw Data: EMPIAR-, EMPIAR-, EMPIAR-
- Marletta Lab
- Britt Lab
Nitric Oxide-Induced Conformational Changes Govern H-NOX and Histidine Kinase Interaction and Regulation in Shewanella oneidensis
Biochemistry
2017
Rao M*, Herzik MA Jr*, Iavarone AT, Marletta MA
Abstract
*These authors contributed equally
Nitric oxide (NO) is implicated in biofilm regulation in several bacterial families via heme-nitric oxide/oxygen binding (H-NOX) protein signaling. Shewanella oneidensis H-NOX (So H-NOX) is associated with a histidine kinase (So HnoK) encoded on the same operon, and together they form a multicomponent signaling network whereby the NO-bound state of So H-NOX inhibits So HnoK autophosphorylation activity, affecting the phosphorylation state of three response regulators. Although the conformational changes of So H-NOX upon NO binding have been structurally characterized, the mechanism of HnoK inhibition by NO-bound So H-NOX remains unclear. In the present study, the molecular details of So H-NOX and So HnoK interaction and regulation are characterized. The N-terminal domain in So HnoK was determined to be the site of H-NOX interaction, and the binding interface on So H-NOX was identified using a combination of hydrogen-deuterium exchange mass spectrometry and surface-scanning mutagenesis. Binding kinetics measurements and analytical gel filtration revealed that NO-bound So H-NOX has a tighter affinity for So HnoK compared that of H-NOX in the unliganded state, correlating binding affinity with kinase inhibition. Kinase activity assays with binding-deficient H-NOX mutants further indicate that while formation of the H-NOX-HnoK complex is required for HnoK to be catalytically active, H-NOX conformational changes upon NO-binding are necessary for HnoK inhibition.
Access the Paper
- PMID: 28170222
- PMCID: Submitted
Additional Links
- PDBs:
- EMDBs:
- Raw Data: EMPIAR-, EMPIAR-, EMPIAR-
- Marletta Lab
Cryo-electron microscopy structure of the TRPV2 ion channel
Nature Structural and Molecular Biology
2016
Zubcevic L*, Herzik MA Jr*, Chung BC, Liu Z, Lander GC, Lee SY
Abstract
*These authors contributed equally
Transient receptor potential vanilloid (TRPV) cation channels are polymodal sensors involved in a variety of physiological processes. TRPV2, a member of the TRPV family, is regulated by temperature, by ligands, such as probenecid and cannabinoids, and by lipids. TRPV2 has been implicated in many biological functions, including somatosensation, osmosensation and innate immunity. Here we present the atomic model of rabbit TRPV2 in its putative desensitized state, as determined by cryo-EM at a nominal resolution of ∼4 Å. In the TRPV2 structure, the transmembrane segment 6 (S6), which is involved in gate opening, adopts a conformation different from the one observed in TRPV1. Structural comparisons of TRPV1 and TRPV2 indicate that a rotation of the ankyrin-repeat domain is coupled to pore opening via the TRP domain, and this pore opening can be modulated by rearrangements in the secondary structure of S6.
Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition
eLIFE
2016
Dambacher CM*, Worden EJ*, Herzik MA Jr*, Martin A, Lander GC
Abstract
*These authors contributed equally
The 26S proteasome is responsible for the selective, ATP-dependent degradation of polyubiquitinated cellular proteins. Removal of ubiquitin chains from targeted substrates at the proteasome is a prerequisite for substrate processing and is accomplished by Rpn11, a deubiquitinase within the 'lid' sub-complex. Prior to the lid's incorporation into the proteasome, Rpn11 deubiquitinase activity is inhibited to prevent unwarranted deubiquitination of polyubiquitinated proteins. Here we present the atomic model of the isolated lid sub-complex, as determined by cryo-electron microscopy at 3.5 Å resolution, revealing how Rpn11 is inhibited through its interaction with a neighboring lid subunit, Rpn5. Through mutagenesis of specific residues, we describe the network of interactions that are required to stabilize this inhibited state. These results provide significant insight into the intricate mechanisms of proteasome assembly, outlining the substantial conformational rearrangements that occur during incorporation of the lid into the 26S holoenzyme, which ultimately activates the deubiquitinase for substrate degradation.
Access the Paper
- PMID: 26744777
- PMCID: PMC4749569
Additional Links
- PDB: 3JCK
- EMDBs:
- Raw Data: EMPIAR-, EMPIAR-, EMPIAR-
- Lander Lab
- Martin Lab
Structural insights into the role of iron-histidine bond cleavage in nitric oxide-induced activation of H-NOX gas sensor proteins
Proceedings of the National Academy of Sciences
2014
Herzik MA Jr, Jonnalagadda R, Kuriyan J, Marletta MA
Abstract
Heme-nitric oxide/oxygen (H-NOX) binding domains are a recently discovered family of heme-based gas sensor proteins that are conserved across eukaryotes and bacteria. Nitric oxide (NO) binding to the heme cofactor of H-NOX proteins has been implicated as a regulatory mechanism for processes ranging from vasodilation in mammals to communal behavior in bacteria. A key molecular event during NO-dependent activation of H-NOX proteins is rupture of the heme-histidine bond and formation of a five-coordinate nitrosyl complex. Although extensive biochemical studies have provided insight into the NO activation mechanism, precise molecular-level details have remained elusive. In the present study, high-resolution crystal structures of the H-NOX protein from Shewanella oneidensis in the unligated, intermediate six-coordinate and activated five-coordinate, NO-bound states are reported. From these structures, it is evident that several structural features in the heme pocket of the unligated protein function to maintain the heme distorted from planarity. NO-induced scission of the iron-histidine bond triggers structural rearrangements in the heme pocket that permit the heme to relax toward planarity, yielding the signaling-competent NO-bound conformation. Here, we also provide characterization of a nonheme metal coordination site occupied by zinc in an H-NOX protein.
Access the Paper
- PMID: 25253889
- PMCID: PMC4210026
Additional Links
- PDBs: 4U99 4U9B 4U9G 4U9J 4U9K
- EMDBs:
- Raw Data: EMPIAR-, EMPIAR-, EMPIAR-
- Marletta Lab
- Kuriyan Lab
Tunnels modulate ligand flux in a heme nitric oxide/oxygen binding (H-NOX) domain
Proceedings of the National Academy of Sciences
2011
Winter MB*, Herzik MA Jr*, Kuriyan J, Marletta MA
Abstract
*These authors contributed equally
Interior topological features, such as pockets and channels, have evolved in proteins to regulate biological functions by facilitating the diffusion of biomolecules. Decades of research using the globins as model heme proteins have clearly highlighted the importance of gas pockets around the heme in controlling the capture and release of O(2). However, much less is known about how ligand migration contributes to the diverse functions of other heme protein scaffolds. Heme nitric oxide/oxygen binding (H-NOX) domains are a conserved family of gas-sensing heme proteins with a divergent fold that are critical to numerous signaling pathways. Utilizing X-ray crystallography with xenon, a tunnel network has been shown to serve as a molecular pathway for ligand diffusion. Structure-guided mutagenesis results show that the tunnels have unexpected effects on gas-sensing properties in H-NOX domains. The findings provide insights on how the flux of biomolecules through protein scaffolds modulates protein chemistry.
Structure and properties of a bis-histidyl ligated globin from Caenorhabditis elegans
Biochemistry
2010
Yoon J, Herzik MA Jr, Winter MB, Tran R, Olea C Jr, Marletta MA
Abstract
Globins are heme-containing proteins that are best known for their roles in oxygen (O(2)) transport and storage. However, more diverse roles of globins in biology are being revealed, including gas and redox sensing. In the nematode Caenorhabditis elegans, 33 globin or globin-like genes were recently identified, some of which are known to be expressed in the sensory neurons of the worm and linked to O(2) sensing behavior. Here, we describe GLB-6, a novel globin-like protein expressed in the neurons of C. elegans. Recombinantly expressed full-length GLB-6 contains a heme site with spectral features that are similar to those of other bis-histidyl ligated globins, such as neuroglobin and cytoglobin. In contrast to these globins, however, ligands such as CO, NO, and CN(-) do not bind to the heme in GLB-6, demonstrating that the endogenous histidine ligands are likely very tightly coordinated. Additionally, GLB-6 exhibits rapid two-state autoxidation kinetics in the presence of physiological O(2) levels as well as a low redox potential (-193 +/- 2 mV). A high-resolution (1.40 A) crystal structure of the ferric form of the heme domain of GLB-6 confirms both the putative globin fold and bis-histidyl ligation and also demonstrates key structural features that can be correlated with the unusual ligand binding and redox properties exhibited by the full-length protein. Taken together, the biochemical properties of GLB-6 suggest that this neural protein would most likely serve as a physiological sensor for O(2) in C. elegans via redox signaling and/or electron transfer.
Access the Paper
- PMID: 20518498
- PMCID: PMC2903878
Additional Links
- PDB: 3MVC
- EMDBs:
- Raw Data: EMPIAR-, EMPIAR-, EMPIAR-
- Marletta Lab
- Kuriyan Lab
Structural insights into the molecular mechanism of H-NOX activation
Protein Science
2010
Olea C Jr, Herzik MA Jr, Kuriyan J, Marletta MA
Abstract
Nitric oxide (NO) signaling in mammals controls important processes such as smooth muscle relaxation and neurotransmission by the activation of soluble guanylate cyclase (sGC). NO binding to the heme domain of sGC leads to dissociation of the iron-histidine (Fe-His) bond, which is required for enzyme activity. The heme domain of sGC belongs to a larger class of proteins called H-NOX (Heme-Nitric oxide/OXygen) binding domains. Previous crystallographic studies on H-NOX domains demonstrate a correlation between heme bending and protein conformation. It was unclear, however, whether these structural changes were important for signal transduction. Subsequent NMR solution structures of H-NOX proteins show a conformational change upon disconnection of the heme and proximal helix, similar to those observed in the crystallographic studies. The atomic details of these conformational changes, however, are lacking in the NMR structures especially at the heme pocket. Here, a high-resolution crystal structure of an H-NOX mutant mimicking a broken Fe-His bond is reported. This mutant exhibits specific changes in heme conformation and major N-terminal displacements relative to the wild-type H-NOX protein. Fe-His ligation is ubiquitous in all H-NOX domains, and therefore, the heme and protein conformational changes observed in this study are likely to occur throughout the H-NOX family when NO binding leads to rupture of the Fe-His bond.
Access the Paper
- PMID: 20162612
- PMCID: PMC2867026
Additional Links
- PDBs: 3LAH 3LAI
- EMDBs:
- Raw Data: EMPIAR-, EMPIAR-, EMPIAR-
- Marletta Lab
- Kuriyan Lab
Spectroscopic Characterization of Successive Phosphorylation of the Tissue Factor Cytoplasmic Region
Open Spectroscopy Journal
2009
Sen M, Herzik MA Jr, Craft JW, Creath AL, Agrawal S, Ruf W, Legge GB
Abstract
Tissue Factor (TF) is well known for its role during the activation of the coagulation pathway, but it is also critical for tumor biology and inflammation through protease activated receptor (PAR) 2 signaling. This signaling function is modulated by the successive phosphorylation of residues Ser253 and Ser258 within the TF cytoplasmic region (TFCR). This paper reports how we used NMR and spectroscopic methods to investigate the structural propensities of the unphosphorylated and phosphorylated forms of the TFCR. When unphosphorylated, the TFCR forms a local hydrophobic collapse around Trp254 and an electropositive patch from the membrane proximal basic block (Arg246-Lys247) to the conserved PKCalpha consensus residue Lys255. Phosphorylation of Ser253 alters the charge characteristics of this membrane proximal region, thereby strengthening the interaction between residue Ala248 and the Trp254 aromatic group. Phosphorylation of the Ser258-Pro259 motif destabilizes a turn at the C-terminus to form an extended polyproline helical motif. Our data suggests that by changing both its charge and local structural propensity, covalent modifications of the TFCR can potentially regulate its association with the cellular membrane and its signaling partners.